

宏基因組測序
宏基因組/宏轉錄組測序
【添加時間:2016-09-20 14:30:31】【來源:】【作者:dggadmin】
宏基因組學這一概念最早是在1998年由威斯康辛大學植物病理學部門的Jo Handelsman等提出的,是源于將來自環境中基因集可以在某種程度上當成一個單個基因組研究分析的想法,而宏的英文是“meta-”,具有更高層組織結構和動態變化的含義。后來加州伯克利分校的Kevin Chen和Lior Pachter將宏基因組定義為“應用現代基因組學的技術直接研究自然狀態下的微生物的有機群落,而不需要在實驗室中分離單一的菌株”的科學。
宏基因組學研究的對象是特定環境中的總DNA,不是某特定的微生物或其細胞中的總DNA,不需要對微生物進行分離培養和純化,這對我們認識和利用未培養微生物提供了一條新的途徑。
宏基因組測序無需PCR擴增,其實驗流程見下圖:
宏基因組測序無需PCR擴增,其實驗流程見下圖:
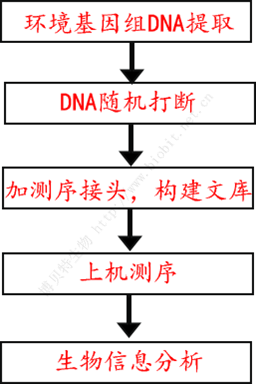
圖1 宏基因組/宏轉錄組實驗流程
宏基因組數據的標準分析流程通常包括質量控制、rRNA識別、序列組裝 (assembly)、基因預測 (gene calling)、基因注釋 (功能和生物來源的annotation) 等步驟(如下圖)。其個性化分析多種多樣,例如多樣性分析(PCoA、NMDS等排序分析、PerMANOVA/Anosim分析)、LEfSE分析、與環境因子的關聯分析 (Mantel test、方差分解分析VPA等)、網絡分析,等等。
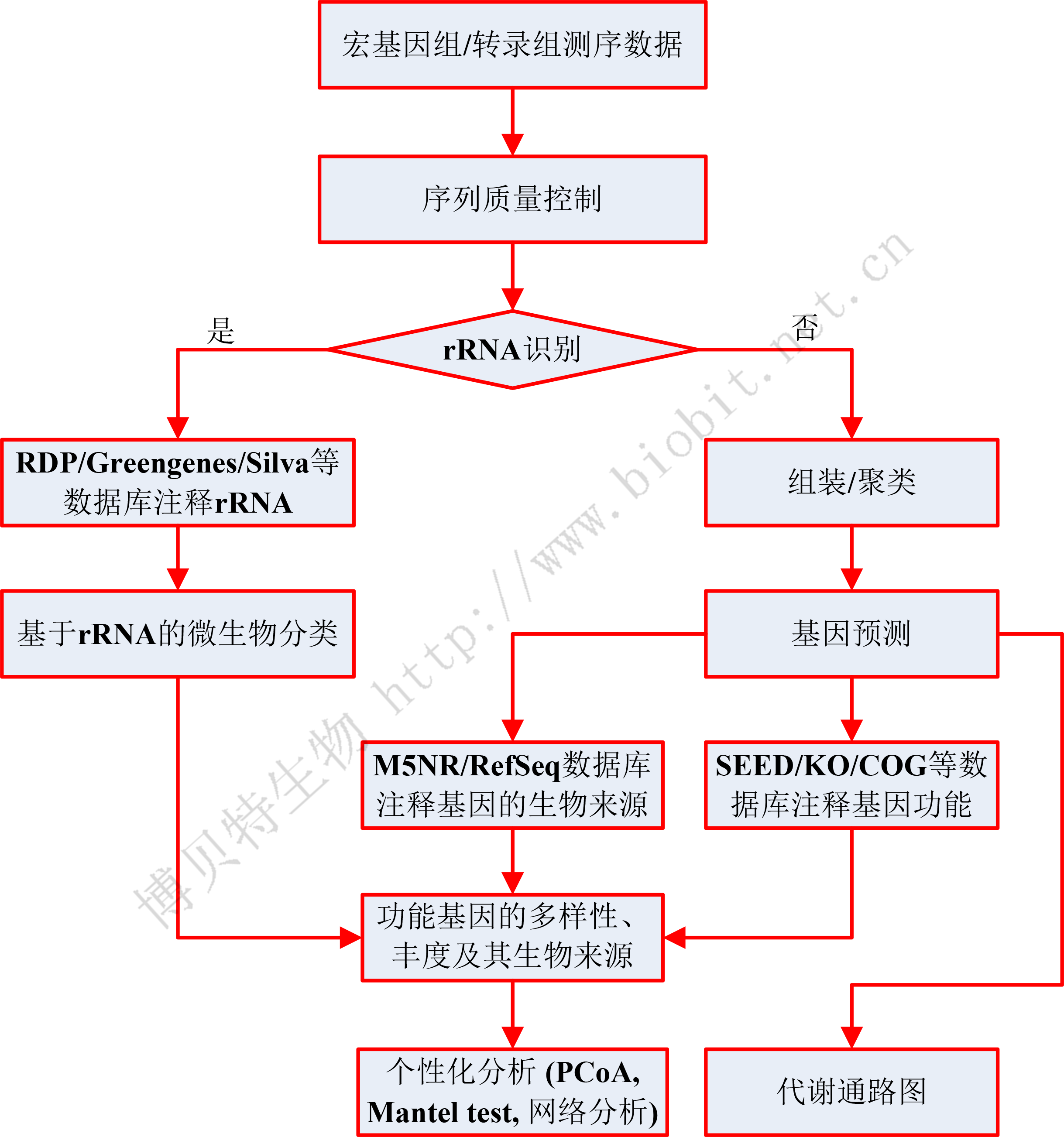
圖2 宏基因組/宏轉錄組生物信息分析流程
宏基因組生物信息分析結果示例如下:
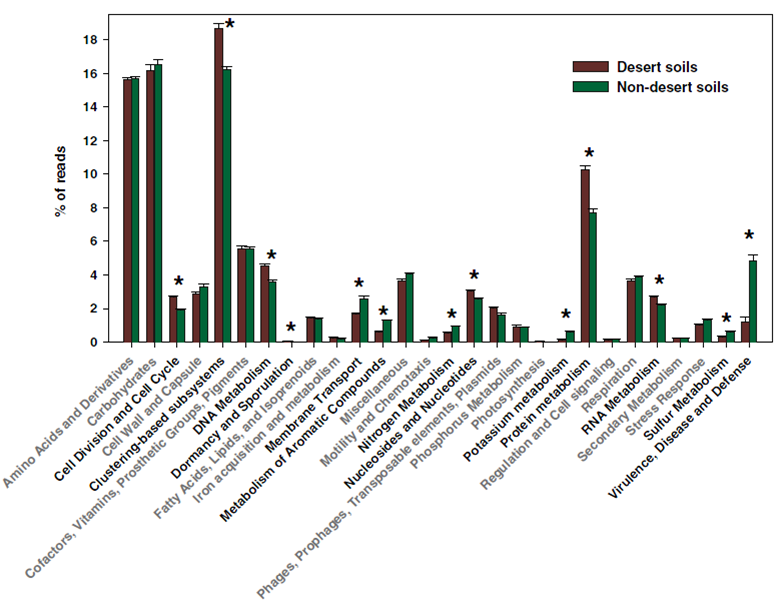
圖3 用柱狀圖呈現不同處理之間功能類別(SEED subsystems功能庫Level 1 水平)的豐度差異(Fierer et al. PNAS 2012)
圖3 用柱狀圖呈現不同處理之間功能類別(SEED subsystems功能庫Level 1 水平)的豐度差異(Fierer et al. PNAS 2012)
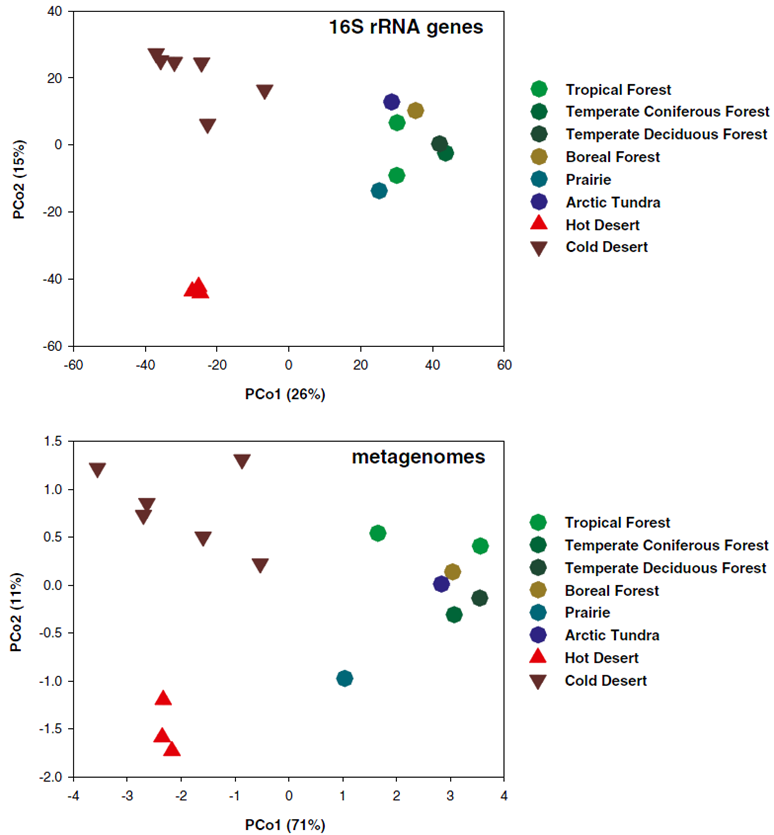
圖4 用PCoA圖呈現微生物群落(a. 16S rRNA)與功能(b. 宏基因組)在beta多樣性上的差異(Fierer et al. PNAS 2012)
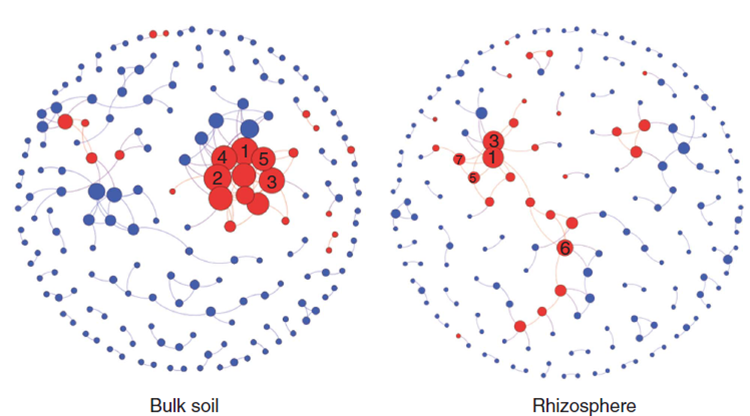
圖5 比較不同處理(非根際土與根際土)之間的微生物功能網絡(Menders et al. ISME J 2014)
圖中藍色節點表示功能,紅色節點為細菌門,連線表示兩節點之間存在顯著相關。
圖中藍色節點表示功能,紅色節點為細菌門,連線表示兩節點之間存在顯著相關。
更多個性化分析及定制服務請咨詢公司客服。
測序訂單下載
